Tag: BryologyPage 1 of 3

It seems that not only are the North American Aneura sharpii, the east Asian Aneura pellioides and the south-east Asian Aneura maxima all distinct species, but there are also two Pellia-like Aneura species in Europe.
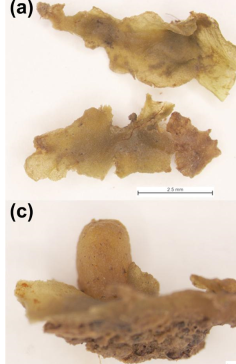
We have chosen a lectoype for Riccardia fuscovirens Lindb. (i.e., a type specimen that is chosen after a name has been published, but from the original specimens or illustrations that the author of the name would have been influenced by when they were first recognizing the taxon)

Geography, and particularly climate, have distinguished the extreme western parts of Scotland from the rest of the country for thousands of years. Many of our rarest plant species…
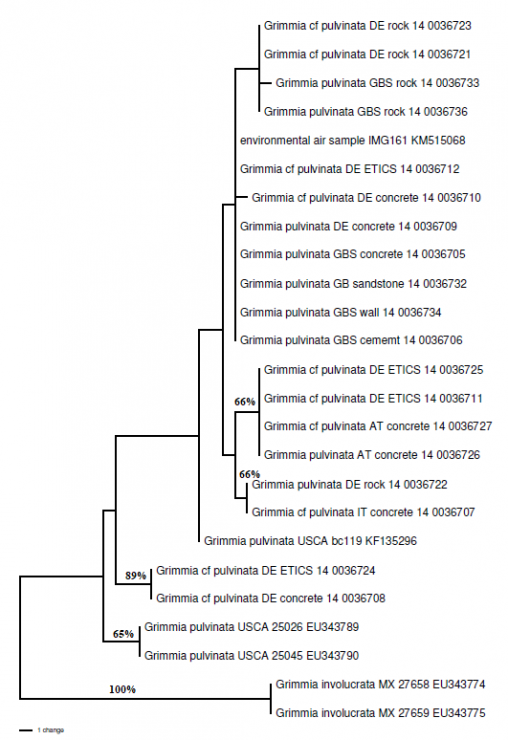
Even with a plant as common, and as commonly overlooked, as this pollution-tolerant urban bryophyte, there is still genetic diversity to explore and explain.
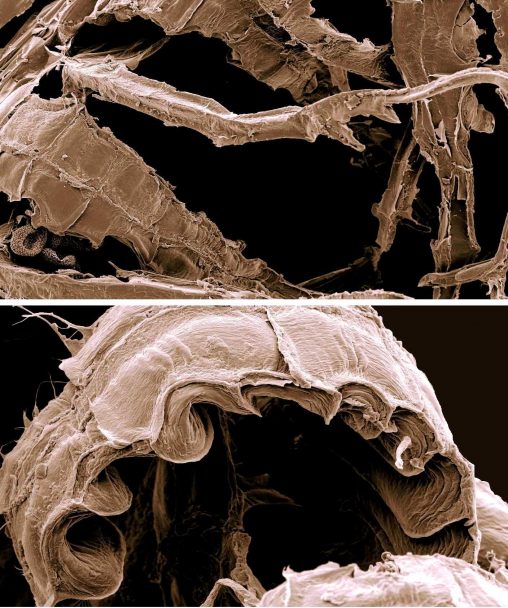
When conservation scientists are trying to decide which species are most in need of protection, the main consideration is usually how likely they are to become extinct, as…
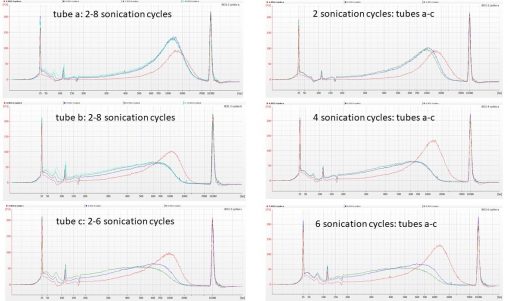
We started our lab work on the Polytrichum hybrid baits project on the 1st of October, by normalising some CTAB-extracted DNA with 0.1X TE to 55 µL of…

The current Next Gen Sequencing lab project at the Botanics involves looking at the phylogeny of Polytrichum section Polytrichum, using hybrid capture. The work will form part of…

There’s an exciting project, The 10KP (10,000 Plants) Genome Sequencing Project, that aims to sequence and characterize representative genomes from every major clade of embryophytes, green algae, and…

Dr Des Callaghan spends rather a lot of his time chasing after rare things. He’s an environmental consultant with many strings to his bow, but a particular specialisation…

The Shawnee National Forest skirts the midwestern town of Carbondale, which is home to one of the campuses of Southern Illinois University. It’s also one of the prettiest…

One of North America’s endemic hornworts, Phaeoceros proskaueri Stotler, Crand.-Stotl. & W.T.Doyle [also known as Paraphymatoceros proskaueri (Stotler, Crand.-Stotl. & W.T.Doyle) J.C.Villarreal & Cargill] was described from plants collected in the Monterey Bay…

During a family holiday to Santiago, Panama in June/July 2011, we snuck in a short bryologising trip, first heading west along the Pan-American Highway, then north, to the…

The Botany 2004 meeting was in Snowbird, Utah – a chance to see a different part of the United States (and, of course, to present our research to…

In April 2004, I flew north from Illinois to met up with a botanical friend, Dr Zoe Badcock. Our meeting point was Vancouver, British Columbia; from there we…

There’s something quite melancholy about going back through all the little paper packets of voucher specimens, remembering who and where you were when you collected them, and thinking…

The Science building at the Botanics closes down between Christmas and New Year, so any last bits of work for the year have to be packed up and…

Now that we have six wild-collected accessions of Plagiochasma currently growing on public display in the RBGE Arid House, from China, the US (Texas) and Saudi Arabia, I’ve…

I’m just back from field work in New Zealand with Yoan Coudert, a French CNRS funded researcher based at the Ecole Normale Supérieure in Lyon. A major objective…

Despite a reputation for being rather a rare breed, this week, purely by chance, we have found ourselves with an embarrassment of bryologists at the Gardens. As well…

Our short damp November days offer the perfect opportunity for leafing through reels of photographs from earlier in the year; many of mine are from a short trip…