Tag: DNA barcodingPage 1 of 2

It seems that not only are the North American Aneura sharpii, the east Asian Aneura pellioides and the south-east Asian Aneura maxima all distinct species, but there are also two Pellia-like Aneura species in Europe.
We have chosen a lectoype for Riccardia fuscovirens Lindb. (i.e., a type specimen that is chosen after a name has been published, but from the original specimens or illustrations that the author of the name would have been influenced by when they were first recognizing the taxon)

The thalloid liverwort Aneura pinguis (L.) Dumort. (basionym Jungermannia pinguis L.) has been reported from a bewildering range of climates and habitats, from neotropical cloud forests to Scottish…

The simple thalloid liverwort Aneura has become a flagship genus for DNA barcoding at RBGE. Only a single widespread species, Aneura pinguis, is traditionally recognized in the UK,…
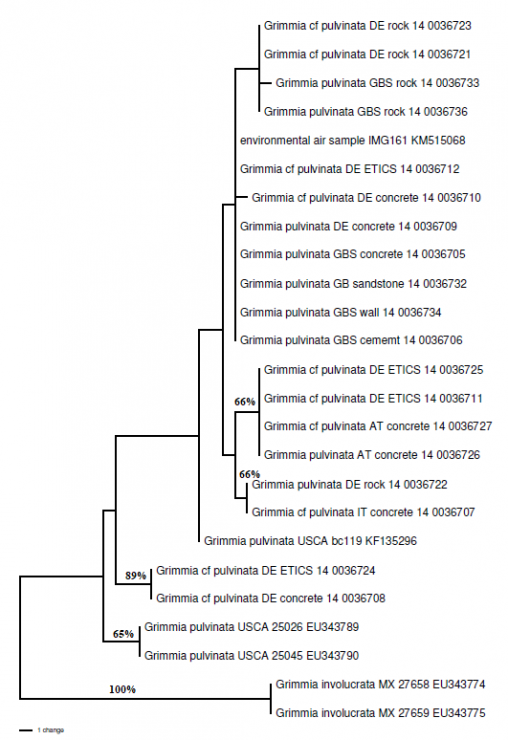
Even with a plant as common, and as commonly overlooked, as this pollution-tolerant urban bryophyte, there is still genetic diversity to explore and explain.

The leafy liverwort genus Nardia has five species that are known to occur in Scotland. Of these five species that occur in Scotland, four are described in Smith’s…

Dr Des Callaghan spends rather a lot of his time chasing after rare things. He’s an environmental consultant with many strings to his bow, but a particular specialisation…

To understand giant panda diet you need to understand bamboos and there are many types of bamboos in their habitat. Giant pandas seems to know which is best…

On a mountain in China a giant panda spends hours sitting eating bamboo but there is no time for a scientist to sit when you are trying to…

After RBGE’s initial involvement in land plant DNA barcode marker selection, culminating in a couple of 2009 papers that both utilized bryophyte barcoding data sets, we started a…

Enigmatic and isolated although it is, it seems that our Australian colleagues have now “got their eye in” for complex thalloid liverwort Monocarpus sphaerocarpus – after many years…

It may only take a mammal 12 seconds to poop – but poo contains a treasure trove of information about the animal and its environment that can take…

Once we realised that most of our plate of Schistidium ITS2 amplifications had been successful, it was an easy decision to process them all for DNA sequencing. If…

Once the polymerase chain reaction is over, it’s time to Run The Gel; this is make-or-break time, when we find out if our PCR amplification has actually worked….

After we extracted a plate’s worth (12 columns by 8 rows, or 96 samples) of Schistidium DNA, the next step in our process is to copy a preselected…

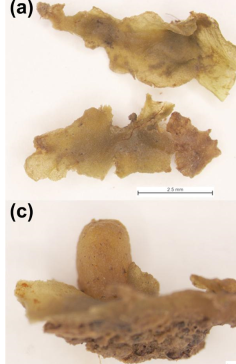
Just over a week into our current Synthesys-funded Schistidium project, and Wolfgang has picked through piles of packets of mosses, selecting the 96 that we would most like…

Monday 27th March was the start of a month-long visit to RBGE by the Fraunhofer Institute for Building Physics‘s Dr Wolfgang Hofbauer, funded by the EU Synthesys Access…

Many new species are already included in natural history collections around the world, it’s just that nobody has yet got around to examining the material, recognising that it represents something…

In conjunction with Dr Daniela Schill’s monographic work on Sphaerocarpos, we’ve been building a molecular phylogeny for the genus. We have attempted to extract DNA from 66 accessions,…

The Sphaerocarpales (or “Bottle Liverworts”) form a very distinct group in the complex thalloid liverworts, with ca. 30 species in five genera: originally the group just included Geothallus…