Tag: moss
Even with a plant as common, and as commonly overlooked, as this pollution-tolerant urban bryophyte, there is still genetic diversity to explore and explain.
When conservation scientists are trying to decide which species are most in need of protection, the main consideration is usually how likely they are to become extinct, as…
We started our lab work on the Polytrichum hybrid baits project on the 1st of October, by normalising some CTAB-extracted DNA with 0.1X TE to 55 µL of…

The current Next Gen Sequencing lab project at the Botanics involves looking at the phylogeny of Polytrichum section Polytrichum, using hybrid capture. The work will form part of…

Dr Des Callaghan spends rather a lot of his time chasing after rare things. He’s an environmental consultant with many strings to his bow, but a particular specialisation…

I’m just back from field work in New Zealand with Yoan Coudert, a French CNRS funded researcher based at the Ecole Normale Supérieure in Lyon. A major objective…

As far as our 2013 RBGE MSc project proposal to generate a phylogeny of Octoblepharum goes, Juan Carlos Villarreal, Noris Salazar Allen and I were clearly not the…

Spring Break’s a big thing in the US, and spring of 2005, Juan Carlos Villarreal and I spent ours on a road-trip down through Louisianna, looking for the…

Several years back, I postdocced in Barbara Crandall-Stotler’s lab in Southern Illinois University, Carbondale. In the late Autumn of 2003, Panamanian bryologist Noris Salazar Allen spent a few…

Once we realised that most of our plate of Schistidium ITS2 amplifications had been successful, it was an easy decision to process them all for DNA sequencing. If…

Once the polymerase chain reaction is over, it’s time to Run The Gel; this is make-or-break time, when we find out if our PCR amplification has actually worked….

After we extracted a plate’s worth (12 columns by 8 rows, or 96 samples) of Schistidium DNA, the next step in our process is to copy a preselected…

Just over a week into our current Synthesys-funded Schistidium project, and Wolfgang has picked through piles of packets of mosses, selecting the 96 that we would most like…

The moss Campylopus introflexus, native to the southern hemisphere, is now considered an invasive plant in parts of Europe and North America. While it occurs on some natural…

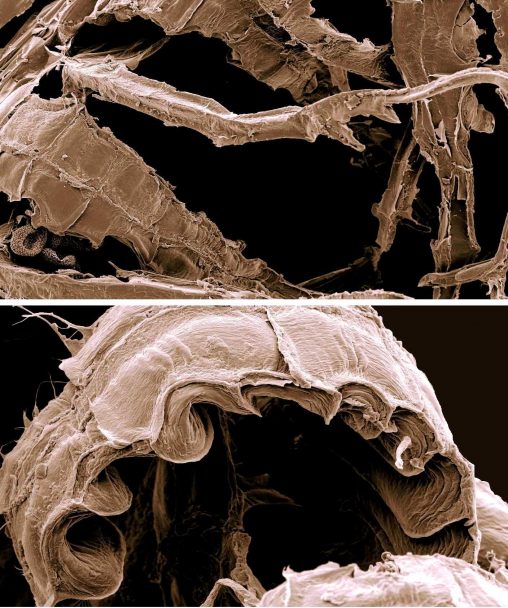
Recently in Kufstein, the home of Austrian bryologist Wolfgang Hofbauer, the demolition of an attractive old building and clearing of trees and other plants from the land, leaving…

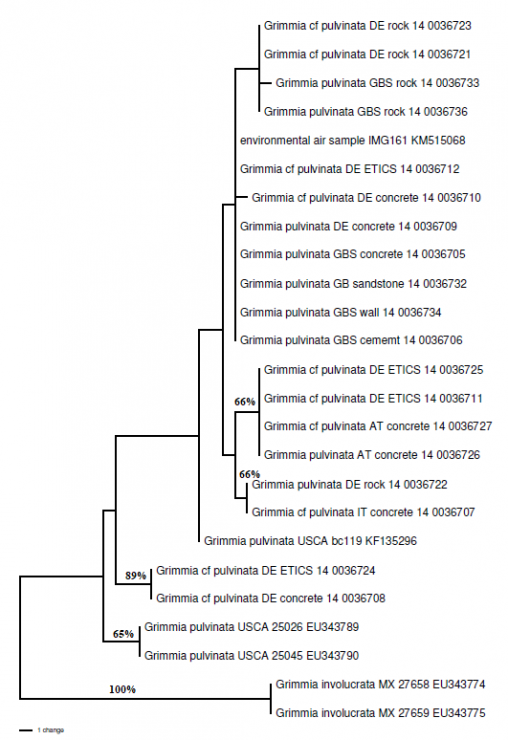
Plant diversity does not have to be far-flung and exotic to be worth studying; even within Scotland, there are unanswered questions about plant distributions. Growing in our towns and…

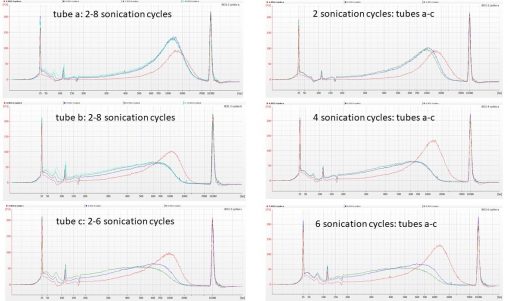
Back in 2014, staff in the molecular lab and herbarium at RBGE greatly enjoyed a three-week visit from Austrian Dr Wolfgang Hofbauer. With funding from the EU SYNTHESYS…

As someone who has used taxpayers’ money to fund research on bryophytes (the collective term for mosses, liverworts and hornworts), ‘But why do bryophytes actually matter?’ is one…