The set of DNA extractions from one of our test plants (#8, Begonia stictopoda RBGE accession 20170115) contained rather low concentrations of DNA. We decided against preparing NGS libraries from these, but instead used the DNA extractions to see if we could successfully concentrate the DNA using a bead clean step, as the volume of DNA extraction that we had for these (80 µl) was greater than the volume we would have needed for the NEBNext® Ultra™ II DNA Library Prep Kit for Ilumina® (60 µl starting volume).

Begonia strictopoda, photographed by Mark Hughes
We used unsonicated DNA, and used the Beckman-Coulter sample purification beads from an Illumina TruSeq Nano DNA kit. We used 1.6x the volume of beads to sample, in 1.5 ml lo-bind tubes, with two washes of 80% ethanol, and eluting in 60 µl of Illumina’s resuspension buffer (RSB).
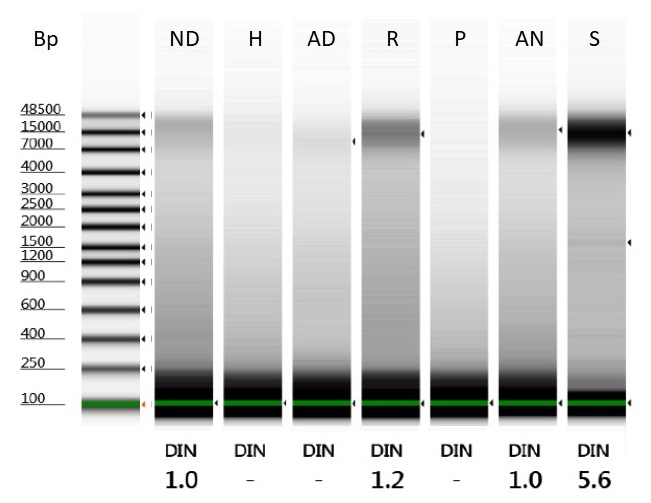
Tapestation image for the genomic DNA extractions from different tissue treatments
We measured DNA concentrations before and after bead-cleaning using a Qubit® dsDNA High Sensitivity Assay kit on a deNovix DS-11 FX, with 1 µl DNA per tube. 1 µl of the extracted DNA was also run on an Agilent Genomic DNA ScreenTape.
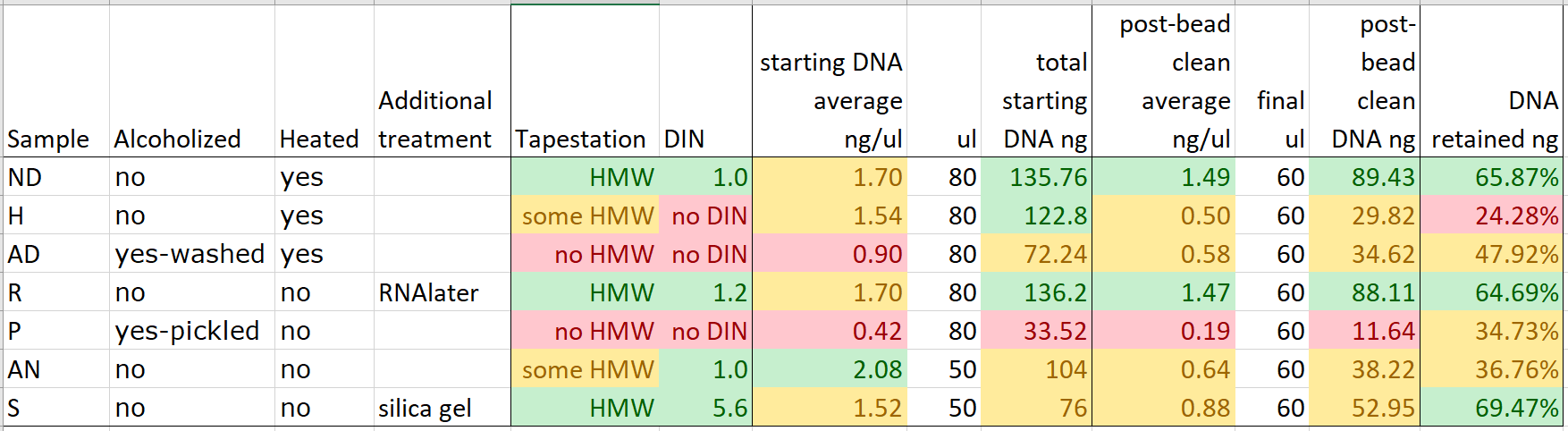
Unfortunately, the bead clean step didn’t concentrate our DNA for any of the extractions, due to the amount of DNA that was lost in each case.
It does seem, though, that while around 30% of the starting DNA was lost during the bead clean step for three of the better-quality DNA extractions (from recently heat-dried herbarium material, RNAlater tissue samples, and silica gel-dried tissue samples), the amount of DNA lost during bead clean rises steeply as the quality of the DNA extractions decreases – for the hairdryer-treated samples, a massive 75% of DNA was lost in the process. This is likely due to an abundance of very small pieces of degraded DNA in some of these extractions.
As far as utilizing highly degraded DNA in library preparation, the implication is that it’s best, if possible, to delay any bead-clean steps until after adaptor ligation.