Tag: molecular labPage 1 of 3

It seems that not only are the North American Aneura sharpii, the east Asian Aneura pellioides and the south-east Asian Aneura maxima all distinct species, but there are also two Pellia-like Aneura species in Europe.
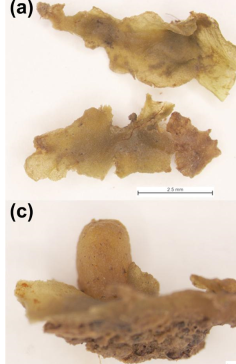
We have chosen a lectoype for Riccardia fuscovirens Lindb. (i.e., a type specimen that is chosen after a name has been published, but from the original specimens or illustrations that the author of the name would have been influenced by when they were first recognizing the taxon)

Our specimen of Aulacomnium androgynum, a moss given the common name “Drumsticks”, was collected for the Darwin Tree of Life project by Dr David Bell on Hatfield Moors, southwest Yorkshire, on the 13th June 2023.

Hylocomiadelphus triquetrus (Rhytidiadelphus triquetrus) has ‘pipe-cleaner moss’ among its many common names. It was collected for the Darwin Tree of Life project by Dr David Bell and Dr Liz Kungu, by the path to the chapel at Dawyk Botanic Garden, on the 1st October 2020.

Arum maculatum – Lords and ladies – was collected for the Darwin Tree of Life project by Dr Maarten Christenhusz (Royal Botanic Garden Kew) on the 27th April 2021, on Petersham common, a conserved woodland in Greater London.

Alchemilla alpina, the alpine Lady’s mantle was collected for the Darwin Tree of Life project by Dr Markus Ruhsam from a rock ledge on Ben Lawers, on the 16th June 2021.

Polygala serpyllifolia – heath milkwort – was collected for the Darwin Tree of Life project by Dr Markus Ruhsam, down from the dam at Lochan na Lairige on Ben Lawers, on the 7th June 2022.

Mnium hornum – Swan’s neck thyme moss – was collected for the Darwin Tree of Life project by Dr David Bell on the 18th August 2020, in the Royal Botanic Garden Edinburgh.

Gooseberry – Ribes uva-crispa – was collected for the Darwin Tree of Life project by Dr Markus Ruhsamn Roslin glen on the 30th April 2023.

Solidago virgaurea – goldenrod – was collected for the Darwin Tree of Life project by Dr Markus Ruhsam in a woodland near the banks of Loch Lomond, on the 1st Sept 2021.

Scapania ornithopoides, the ‘Birds-foot Earwort’ was collected for the Darwin Tree of Life project by Dr David Bell and Dr David Long at the Beinn Eighe Nature Reserve on the 22nd August 2021.

Orchis mascula was collected for the Darwin Tree of Life project by Dr Markus Ruhsam on the 23rd May 2023, near Lochan na Lairige on Ben Lawers. One of the Scots names for this plant is Hen’s kames

Geranium molle, the Dove’s foot cranebill, was collected for the Darwin Tree of Life project on the 9th of May 2022 by Dr Markus Ruhsam, on the verge of a road that passes over a golf course.

A specimen of Pyrus communis, the pear tree, was collected for the DToL project by Dr Markus Ruhsam at the Hermitage of Braid in Edinburgh on the 31st May 2022.

We have already seen the release of the thousandth Darwin Tree of Life genome, the Purple Bar moth, Cosmorhoe ocellata. To celebrate this festive season, we have considered what we have given, or might like to receive, for our own twelve days of Christmas…

The thalloid liverwort Aneura pinguis (L.) Dumort. (basionym Jungermannia pinguis L.) has been reported from a bewildering range of climates and habitats, from neotropical cloud forests to Scottish…
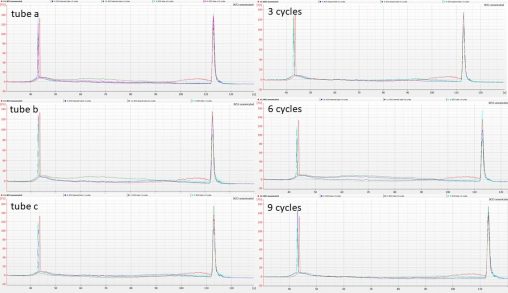
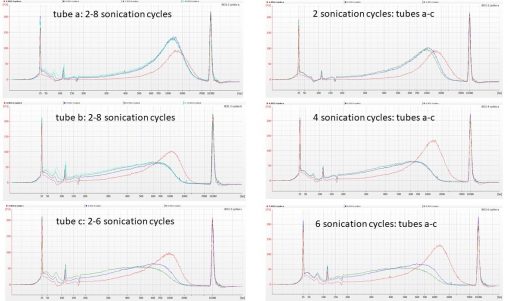
Following on from the rather unpredictable results we obtained from fragmenting duplicate aliquots of CTAB-extracted Polytrichum DNA in the Bioruptor, Isuru cleaned aliquots of IK31 and IK53 using…
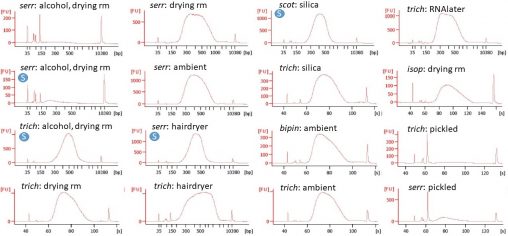
When we’re working out a protocol or troubleshooting, we spend a lot of time quantifying small quantities of fluids, looking at DNA concentrations on the DeNovix, running tapes…
We started our lab work on the Polytrichum hybrid baits project on the 1st of October, by normalising some CTAB-extracted DNA with 0.1X TE to 55 µL of…

The current Next Gen Sequencing lab project at the Botanics involves looking at the phylogeny of Polytrichum section Polytrichum, using hybrid capture. The work will form part of…