When we’re working out a protocol or troubleshooting, we spend a lot of time quantifying small quantities of fluids, looking at DNA concentrations on the DeNovix, running tapes on the Tapestation or chips on the Bioanalyser.
At some early point in the Begonia hybrid capture project, we were on track to generate lots of libraries from a range of plants and treatments. We were working to a deadline, without a huge amount of wriggle-room, but in general it was looking okay. After all, we’d already dealt with the sampling and extracted all the DNA we needed, it was well quantified, and we were heading into some rather routine library preps.
(Spoiler alert: There’s nothing routine about lab work. There’s always at least one spanner you didn’t see coming. We totally missed the deadline.)
Anyway, we were in the lab making the first 16 libraries, using the NEBNext Ultra library preparation kit for Illumina kits (E7370S).
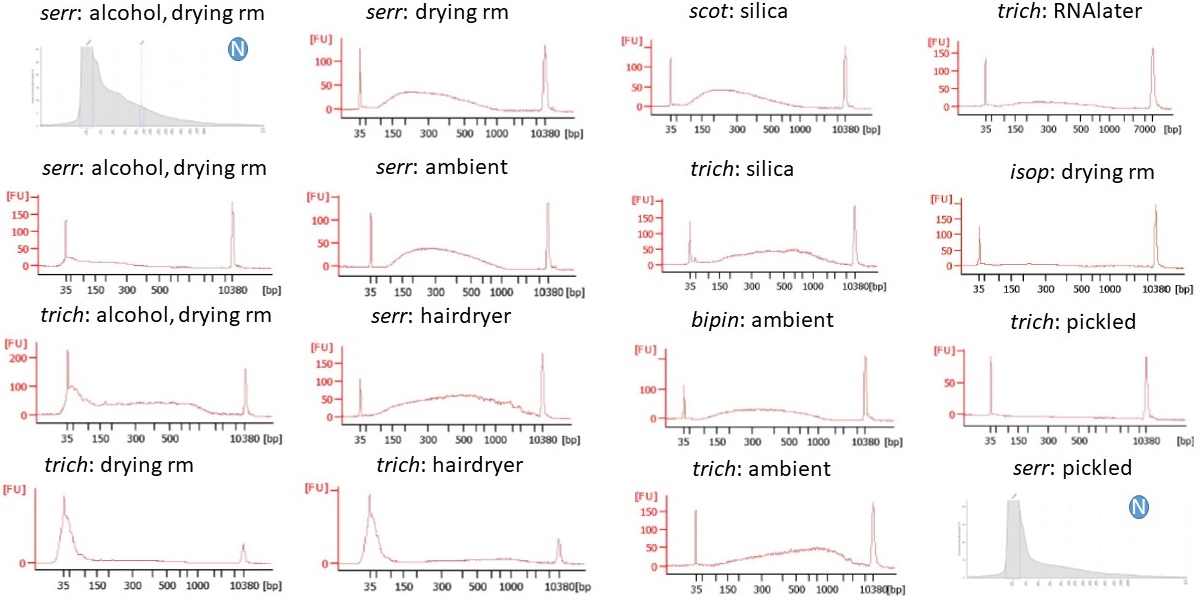
Begonia DNA used in the libraries and run on a Bioanalyser High Sensitivity kit. A circled “N” indicates that the DNA was not sonicated; those two samples were not run on the Bioanalyser, but the Agilent Genomic DNA ScreenTape profiles are shown instead.
We’d started off with between 30 and 160 ng of DNA per library, sonicated all but two of them for 2 or more cycles, size selected four, bead cleaned the rest, normalised so that around 60 ng of DNA went into the end repair, stuck on adaptors, bead cleaned again, quantified the libraries, amplified them with PCR (attaching the indices at this point), bead cleaned again, requantified… and that’s when we discovered that some of our libraries were effectively missing…
| Begonia species | treatment | ng DNA used for Library | size selection | ng DNA post-size selection / bead clean | no. PCR cycles | ng DNA post-PCR / bead clean | % DNA increase after PCR |
| serratipetala | Alcohol, heat | 159.5 | No | 52.80 | 6 | 17.6 | -67 |
| serratipetala | Alcohol, heat | 159.5 | Yes | 9.15 | 9 | 15.1 | 65 |
| trichopoda | Alcohol, heat | 119.8 | Yes | 10.20 | 9 | 255.5 | 2404 |
| trichopoda | Drying room | 119.0 | No | 24.30 | 9 | 687.0 | 2727 |
| serratipetala | Drying room | 112.0 | No | 58.20 | 6 | 381.0 | 555 |
| serratipetala | Ambient | 109.0 | No | 67.50 | 6 | 527.8 | 682 |
| serratipetala | Hairdryer | 100.3 | Yes | 10.20 | 9 | 450.0 | 4312 |
| trichopoda | Hairdryer | 95.5 | No | 18.15 | 9 | 267.6 | 1374 |
| scottii | Silica gel dried | 90.5 | Yes | 12.98 | 9 | 420.0 | 3137 |
| trichopoda | Silica gel dried | 85.0 | No | 24.30 | 9 | 927.0 | 3715 |
| bipinnatifida | Ambient | 78.3 | No | 39.53 | 9 | 802.5 | 1930 |
| trichopoda | Ambient | 66.3 | No | 21.15 | 9 | 1138.5 | 5283 |
| trichopoda | Hairdryer | 64.8 | No | 20.93 | 9 | 760.5 | 3534 |
| isoptera | Drying room | 53.5 | No | 39.23 | 9 | 305.2 | 678 |
| trichopoda | Pickled | 35.3 | No | 2.85 | 10 | 20.0 | 601 |
| serratipetala | Pickled | 34.6 | No | 3.38 | 10 | 54.3 | 1509 |
The two libraries that we were creating from Begonia serratipetala tissue that had been treated with alcohol and dried in the RBGE drying room had had the highest starting DNA concentrations of any of the treatments. One of these went through size selection, so a far smaller amount of DNA went into the post-amplification PCR, but there was certaintly a considerable amount of DNA (c. 53 ng) going into the PCR for the unselected library.
And we lost it. We did a PCR, followed by a bead clean, and we quantified our DNA expecting to have more than we’d put into the reaction (because, well, it’s PCR….) – and found that in that particular library we ended up with 67% LESS DNA than we’d put into the PCR tube.
To be fair, that was the most extreme example. For the size-selected tube we did get a slight increase, of 65% – hardly the sort of exponential rise you might expect after 9 PCR cycles.

Begonia libraries after PCR enrichment, run on a Bioanalyser High Sensitivity kit. The circled “S” on four of the traces indicates that the DNA fragments were size-selected.
This was a bit of a “back to the drawing-board” moment, as we wondered what had gone wrong, and started to think about DNA repair. In a previous study on hybrid capture of libraries constructed from herbarium material of the legume Inga (Hart et al. 2016), we hadn’t actually seen much difference between DNA that had been repaired or not, and so for this Begonia study, we didn’t use a repair step. Skipping repair saves time, money, and also potentially DNA, as we lost a lot of our repaired Inga DNA. But with Inga, we hadn’t been working with anything that had been treated with alcohol – so was it possible that alcohol damage was part of the problem with our Begonia material?
We had been quantifying our DNA and libraries at multiple steps. The Begonia serratipetala samples that had been treated with alcohol didn’t have particularly unusual tapestation fragment distribution profiles, they still contained DNA after bead cleans, end prep and adaptor ligation, so what was the issue with the PCR..? The most obvious answer seems to be heat. Up until the PCR, the DNA was never heated above 65°C (for 30 minutes, for the library End Prep), whereas the PCR enrichment of the adaptor-ligated DNA includes regular denaturing of the double-stranded DNA at 98°C.
At this point, double-stranded fragments of DNA that contain nicks can separate into several pieces. Any “broken” pieces won’t incorporate adaptors at their ends and won’t amplify. And any DNA fragments that are less than c. 150 bp will be subsequently lost during the post-amplification bead-clean. Hence, we can end up with less DNA than we put in.
While it’s quite nice to come up with an explanation for an annoying result, at this point, instead of pushing ahead with the hybrid capture and a MiSeq run on these samples, we stepped back and reconsidered the experimental design of our project in a way that factored in a DNA repair step.
(And this is one of several cascading reasons why we managed to completely miss an August manuscript deadline….)