A few days ago, I read a tweet from the Botany2015 meeting in Alberta that described DNA extracted from herbarium specimens as “pre-sheared”. This resonates with our own experiences with Inga, where herbarium DNA required very little, if any, fragmentation. However, this is only part of the library preparation; we optimized other parts of the protocol to take account of the degraded nature of the herbarium DNA samples.
With an estimated 30% of starting DNA lost by the end of the first bead-clean up step in the Illumina Tru-Seq Nano protocol (James Nicholls pers. commun.), for DNA that did not need sheared and cleaned (the “pre-sheared” herbarium material) we reduced the starting quantity of genomic DNA for the Tru-Seq kits to 70 ng. We repaired the ends of the DNA fragments (i.e. generated blunt-end DNA fragments) using a combination of fill-in reactions and exonuclease activity, again following the protocol. This was followed by the size selection step, where, having moved heaven and earth to extract enough DNA for your next-generation sequencing method, you proceed to thow away all the bits of DNA that are longer or shorter than a certain size.
For the Tru-Seq libraries that we were preparing, we used Illumina’s Sample Purification Beads for size selection, following the manufacturer’s protocol, aiming for an average insert size of c. 350 bp.
Having extracted a huge amount of DNA from one of our herbarium sheets (1932 flower), we were able to test our size selection on five aliquots of an extraction, which had a Tapestation fragment size distribution of 38 to 937 bp. This was unsonicated DNA, “pre-sheared” by degradation; our test aliquots each comprised 72 ng of DNA in 100 μl.
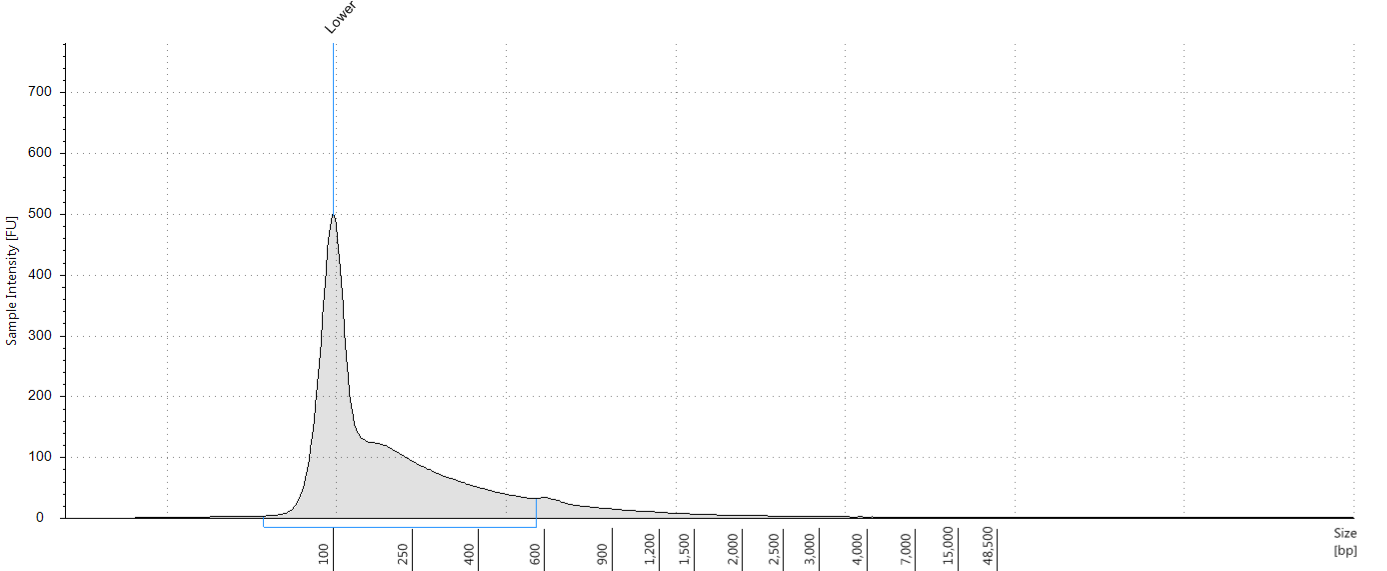
Tapestation trace for DNA extracted from 1932 Inga umbellifera flower, after DNA repair

Bioanalyser trace for DNA extracted from 1932 Inga umbellifera flower, after DNA repair
The first step in the size selection involves using a diluted bead solution, to get rid of large DNA fragments following the Tru-Seq protocol for 350 bp libraries; these stick to the beads and are thus removed from solution. The second step uses undiluted beads, to get rid of really small bits of DNA. In order to remove all small fragments (below c. 100 bp) from our libraries, we trialed the following five treatments for this second step:
a. 30 μl undiluted beads (resulted in peak at 600 bp)
b. 40 μl undiluted beads (resulted in peak at 500 bp)
c. 50 μl undiluted beads (resulted in peak at 400 bp)
d. 60 μl undiluted beads (resulted in peak at 330 bp)
e. 70 μl undiluted beads (resulted in peak at 300 bp)
Bioanalyser traces for all five of these size selection tests, on the same starting DNA, are shown below.
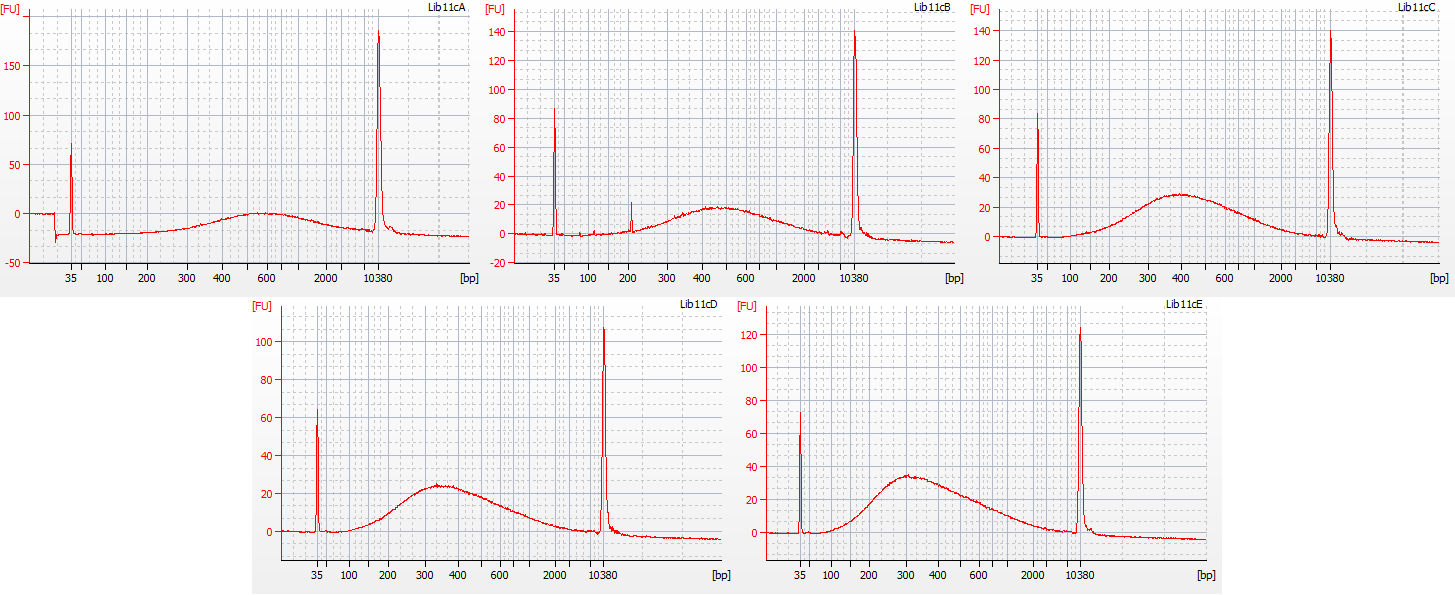
Bioanalyser traces for the five size selection tests on aliquots of the 1932 flower DNA
All five treatments resulted in the loss of DNA fragments shorter than 100 bp. The third treatment, with 50 μl undiluted beads, gave a good distribution, with the peak height around 400 bp – however, it lost a greater proportion of the smaller DNA fragments compared to the fifth treatment (70 μl undiluted beads), which had a mean fragment size of 300 bp. We decided to continue with library preparation using both of these size selection treatments, to see how much information may be lost by ‘throwing away’ more of the smaller DNA fragments.
The results of size selection on the libraries that we used for the rest of the process are shown in the bioanalyser traces below:
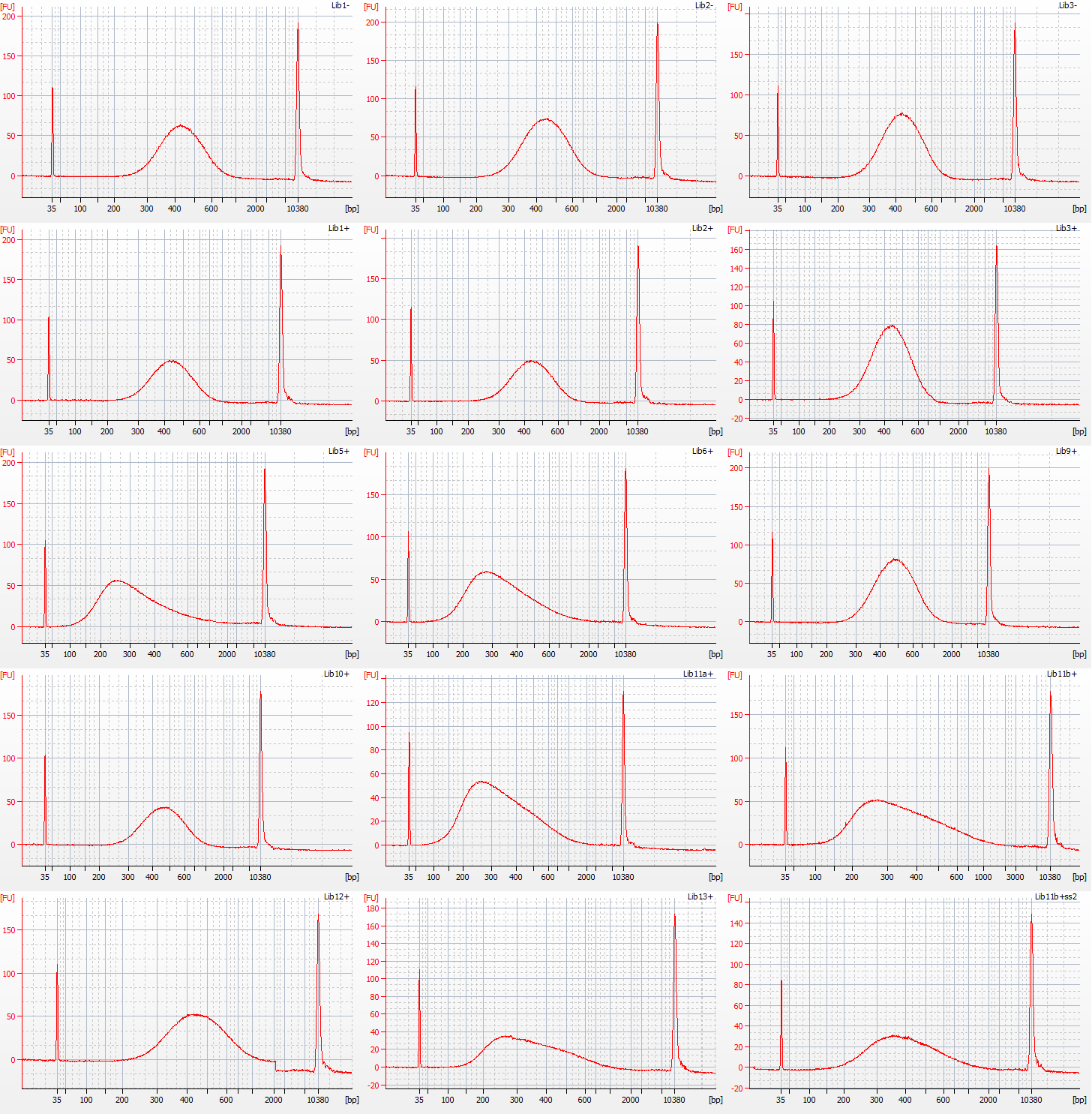
Bioanalyser traces for size selected Inga umbellifera libraries: top row – unrepaired DNA: 2009 herbarium; 2009 silica; 2009 silica; repaired DNA: second row – 2009 herbarium, 2009 silica, 2009 herbarium; third row – 1948; 1948; 2004 herbarium; fourth row – 2004 herbarium; 1932 flower; 1932 flower; bottom row – 1932 leaf; 1835; 1932 flower size select 2
The most recent material, from 2004 and 2009 silica and herbarium material, has DNA fragment size peaks around 400 bp, while the older herbarium material tends to peak between 250-350 bp. The most notable exception is the extraction from the 1932 leaf material, with a peak at over 400 bp. One other 1932 library, generated using modified size selection, also has a less left-skewed fragment distribution, with a peak around 400 bp.
For the NEB libraries, we used two different size-selection protocols. For the DNA from 2009, 2004 and the 1932 leaf material, we followed the protocols for a 400-500 bp insert. For the DNA from the 1932 flower material, and the 1948 and 1835 leaf material, we followed the protocols for a 250-300 bp insert.
Two NEB libraries were generated from 1840 herbarium material, without any size selection – there was too little DNA in the first place to risk losing it!
James A. Nicholls, R. Toby Pennington, Erik J.M. Koenen, Colin E. Hughes, Jack Hearn, Lynsey Bunnefeld, Kyle G. Dexter, Graham N. Stone & Catherine A. Kidner. 2015. Using targeted enrichment of nuclear genes to increase phylogenetic resolution in the neotropical rain forest genus Inga (Leguminosae: Mimosoideae). Frontiers in Plant Science 6: 710. doi: 10.3389/fpls.2015.00710
Capturing Genes from Herbaria. I. What it’s all about. http://stories.rbge.org.uk/archives/16411
Capturing Genes from Herbaria. II. Inga. http://stories.rbge.org.uk/archives/16427
Capturing Genes from Herbaria. III. The samples. http://stories.rbge.org.uk/archives/16441
Capturing Genes from Herbaria. IV. DNA. http://stories.rbge.org.uk/archives/16470
Capturing Genes from Herbaria. V. Fragmenting the DNA. http://stories.rbge.org.uk/archives/16525
Capturing Genes from Herbaria. VI. Size Selection. http://stories.rbge.org.uk/archives/16645
Capturing Genes from Herbaria. VII. Comparisons. http://stories.rbge.org.uk/archives/16737
Capturing Genes from Herbaria. VIII. Amplification. http://stories.rbge.org.uk/archives/16788
Capturing Genes from Herbaria. IX. Hybrid capture. http://stories.rbge.org.uk/archives/17298
Capturing Genes from Herbaria. X. An update. http://stories.rbge.org.uk/archives/20751
Capturing Genes from Herbaria. XI. Some metagenomics of a herbarium specimen. http://stories.rbge.org.uk/archives/20817