It’s a confusing world out there – betaine, DMSO, bovine serum albumin (BSA), trehalose, glycerol, formamide – the list of things that you can throw into a PCR to make it work better (or work at all) is long, and to make it even worse, there’s no need to stick to one additive; sometimes two or more can make the difference between getting no bands and getting bands on your gel. Then, of course, you can vary the amount of your selected additive/s (and when you’re done with that, you can vary the amount of magnesium, or polymerase, or template DNA, or all of them, and then you can vary the reaction times and temperatures; in fact, it would probably be possible to spend the rest of your life just optimizing a single PCR, if you were so inclined).
The additives themselves are a little like mops and irons. The mops (e.g. BSA, Tween-20) mop up ‘bad stuff’ that you’ve inadvertently added to your PCR mix along with your genomic DNA, and the irons (e.g. DMSO, betaine, trehalose) weaken bonds holding DNA together and straighten out DNA that has strong secondary structure, so that the polymerase enzyme can get in and work better. Although this applies particularly to loci like the nuclear ribosomal regions, where the DNA sequences are full of the stronger G-C bonds, even plastid regions that are not considered to have very strong secondary structure and are biased towards weaker A-T bonds often amplify better if you throw a few ‘irons’ into the mix.
At RBGE something called Combinatorial Enhancer Solution (CES) has become one of the most popular additives. The recipe we use contains betaine, DMSO and BSA, while Ralser et al.’s original published recipe also includes dithiothreitol (DTT). For many loci our default PCR recipe involves adding the pertient amount of a 5X CES stock and our success rates from these are usually rather good. However a couple of months ago a new enhancer solution, TBT-PAR, came to our attention. This solution contains trehalose, BSA, Tween-20 and a bit of Tris-HCl, and we have subsequently handed out trial aliquots to any of our lab users who are prepared to give it a go.
And how have we found it? Well, while it would be lovely to present a nice scientific study with replicates and statistical significance, showing the relative benefits of one additive over another, the more I thought about it, the more complicated it seemed – we already know that some additives work better for some loci, and other additives work better for others – so which loci should we try to amplify? Some additives also seem to work better for some taxa than for others, so how many taxa should be included? Even for a standard PCR, the amount of magnesium and the cycling conditions can also make so much difference – should these be tested too? Of course one would need more than one replicate per sample. Lastly, it’s not enough to just see bands on a gel to know whether the additive is successful; everyone who has put in their time at the gel-front will have either experienced or heard anecdotes about beautiful bright bands that simply refuse to sequence, and this is often blamed on PCR additives. Therefore each band should be Sanger sequenced in at least one direction. So, by now we’re talking about the sort of major project that eats up time and money, and recommendations resulting from which may only be applicable to one particular taxon for one particular locus. As you can probably tell, I quickly talked myself out of even starting.
However, that’s not quite the end of the story. What I have done is taken a set of Nepeta (catmint) DNAs from RBGE Honours‘ student Sarah Carlton’s work. These were extracted from herbarium samples of various ages (from 1931 to 2012), and compounding the problems of degraded DNA in this sort of material, mints and their relatives are known to contain secondary compounds that can inhibit PCR. We had initially tried to amplify these samples for the nuclear ITS region using the CES additive, with low success (there were bands for 12 out of 24 samples, but full length good quality sequences were only generated for 3 of these). Using the PCR product from this initial amplification for a second round of ‘nested PCR’ gave us many more bands – but on sequencing it became clear that a proportion of these were due to contamination and not from Nepeta at all, which then cast doubt on other sequences generated the same way (perhaps they were Nepeta, but there was no way to be sure they were the right Nepeta). As a last-ditch effort, I repeated the reactions using TBT-PAR instead of CES. The PCR success rate shot up, and good quality sequences were generated for 18 of the samples (including one from 1931 and two from 1952).
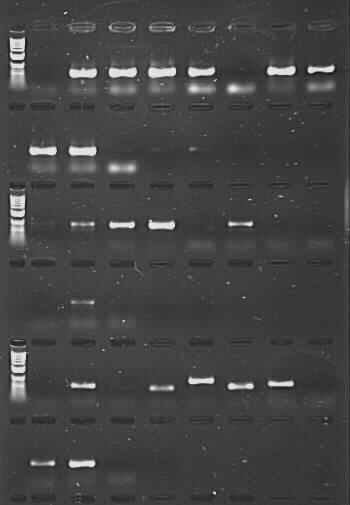
PCR amplification of herbarium DNA using TBT-PAR additive
We routinely generate molecular data from a huge taxonomic array of organisms, including flowering plants, conifers, ferns, liverworts, hornworts, mosses, diatoms, the fungal and algal partners in lichens, rusts and other fungi. Using TBT-PAR I have obtained sequences from Gesneriaceae (for nuclear ITS and plastid spacer psbA-trnH) and Fossombroniaceae (for plastid gene rbcL) that did not amplify using CES and have also heard promising noises from colleagues working on lichens, mosses and Begonia , who are now getting PCR products from DNA that was previously unamplifiable. On the other hand, for Castratella (Melastomataceae) it seems as if PCR success is similar whether CES or TBT-PAR is added.
So, my unscientific gut-feeling-for-now advice for sub-optimal plant DNA is currently: sling some TBT-PAR into the mix first time around, and if that fails, give CES a go – unless, of course, you have another methodology that works better for you…
See also:
JCarlos Villarreal
Pretty useful!!
thanks!